
DEVIATION HANDLING
Created By Ariyono W Ardi , Drs. Apt. MM .on 24 Oct 2022
Penyimpangan (Deviation)
- Unexpected events that occur during the operation, documentation, receipt and storage processes (raw materials, packaging materials, intermediate products and finished products), the manufacturing process, the analysis and distribution of medicinal products that are not in accordance with standard operating procedures and/or specified specifications
- Failure to follow SOPs or meet established specifications
- Deviations must be reported immediately when they occur and analyzed to assess their impact
- Deviations are also known as non-conformities (Non-conformity/ Non-conformance)
Scope of Deviations ( Deviation)
Scope of Deviation Deviation related to:
- Limits and specifications
- Error in implementing c-GMP
- Reprocessing/ Rework
- Unapproved changes
- Implementation of GMP activities without adequate training
- Process parameter/ In-process control beyond the limit
- Failure to implement the procedure correctly.
Deviation Contributor
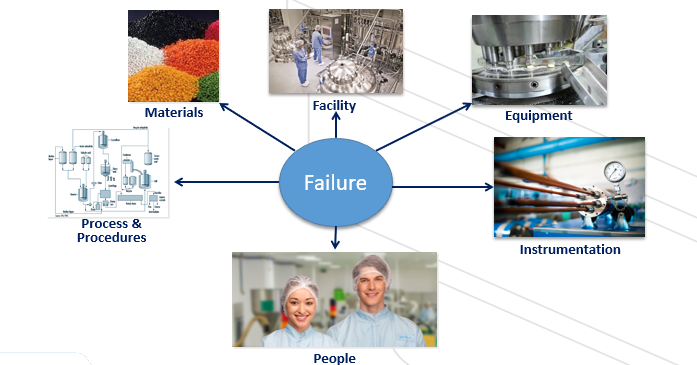
Types of Deviations
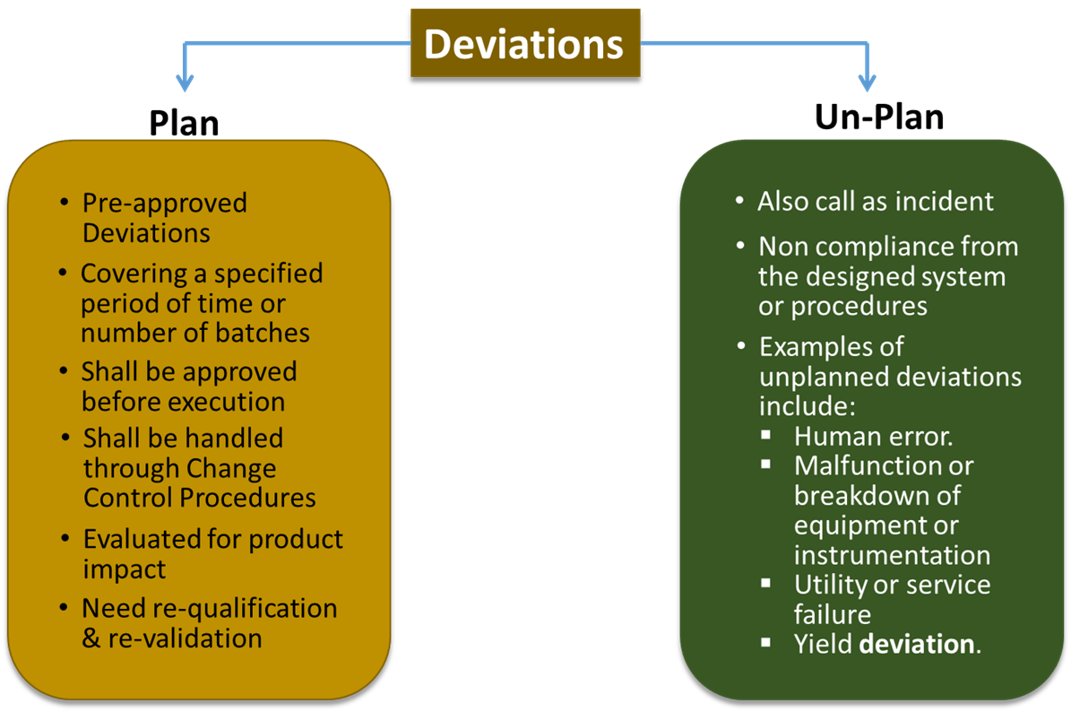
In general, deviations are grouped into 2 (two) groups:
- Planned Deviation.
- Planned deviations are temporary deviations from existing protocols or processes where these deviations must be approved before being implemented
- Planned before implementation with adequate scientific justification.
- It is only allowed to be used within a certain time span or for a certain number of batches.
- This planned deviation may have an impact on the quality, safety, and efficacy of the medicinal product
Example :
- Reducing batch size due to limited raw materials
- Changes in supplier excipients
- Delaying calibration or validation for some reason
Deviation Process Flow

- Un-planned Deviation
- Unplanned deviation is non-compliance with agreed protocols or systems that occurs at any stage of the product life cycle, including manufacturing, testing, packaging and storage.
- Unplanned deviations are also referred to as unplanned events, or uncontrollable events.
- Can be caused by human error, utility failure, damage or malfunction of the machine/instrument.
Example:
-
- Disturbance of the power source resulting in engine trouble
- Machine failure due to personnel error
- Interruption to HVAC or other utility facilities
Un-planned Deviation Process Flow
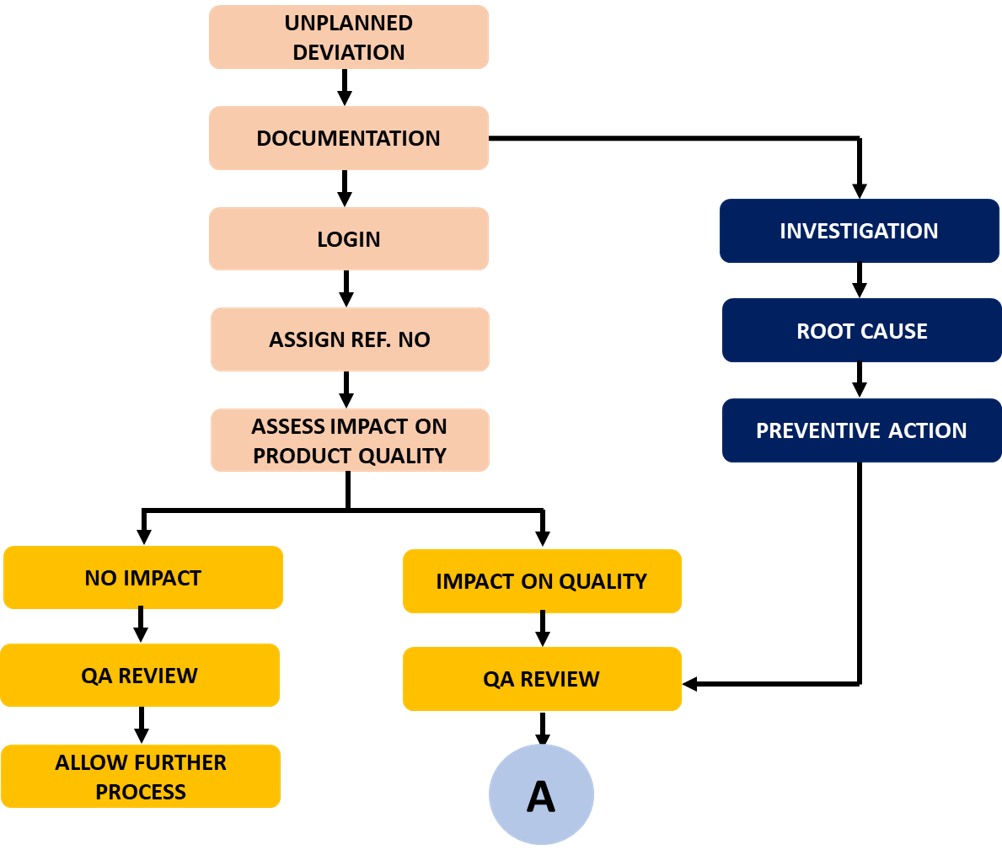
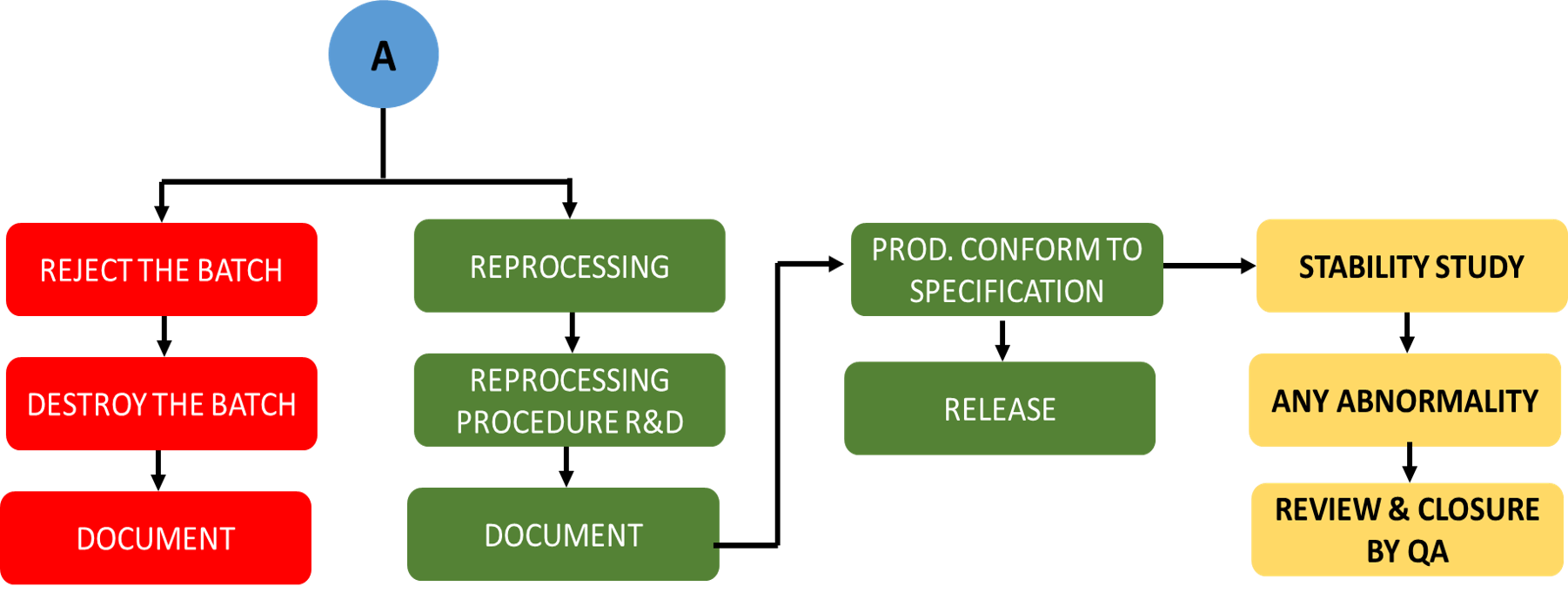
Un-planned Deviation Process Flow
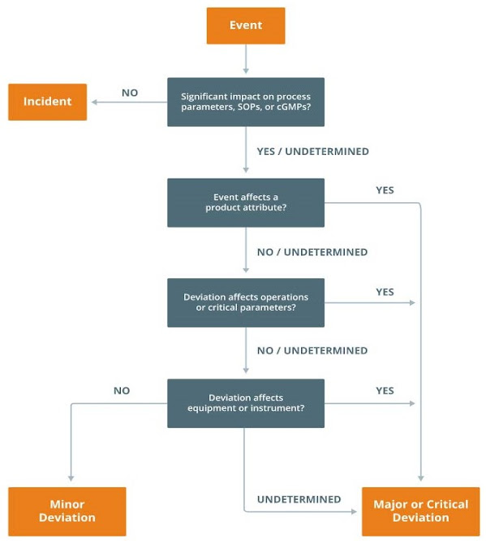
Deviation Classification
- Incident
A simple risk assessment assessment to define “Incident” deviations is to answer the following questions when a deviation is encountered:
- Will the deviation affect product quality attributes, operational parameters, or product quality?
- Does the deviation contradict or omit a requirement in an approved written procedure or specification?
If the answer is NO to questions a) and b) above, the deviation can be considered an Incident (event that is irrelevant and does not affect product quality)
Deviations that do not have a direct impact on product quality, even though this deviation violates the GMP rules (c- GMP)
Example:
- Unauthorized personnel in the production area
- Raw materials spill in the production area
- Personnel wearing gowning in the production room that does not comply with Standard Operating Procedures
- Minor
deviations that do not affect quality attributes, critical process parameters, or equipment or instruments critical to the process or control
Example:
- Minor errors in batch records or other quality documents that have no impact on data integrity.
- Spillage of raw materials during the weighing/dispensing process
- Failure to meet environmental conditions as determined during batch processing.
- The use of the FEFO (First Expired First Out) concept in material handling.
- Use of out-of-tolerance scales to weigh raw materials upon receipt at the warehouse
- Major
Deviations that can have a moderate impact on product quality or the GMP System
Example:
- Machine failure during batch processing
- Mix-up cartons of the same product with different strengths
- Use of unapproved reference standard to test an API or drug product.
- Inadequately trained personnel to perform sterility tests.
- Production started without line clearance.
- Filter integrity test has been carried out using equipment with no documented installation qualification completed.
- Gross misbehavior of staff in a critical aseptic process.
- Pressure differential out of established limits in aseptic fill areas.
- Operational parameter out of range for a parameter defined as non-critical.
- Untrained personnel responsible for segregating the approved and rejected raw material in the warehouse
- Critical
Deviations that have a critical impact on product quality or GMP systems
Example:
- Cross-contamination or product mix-ups
- Process failure during batch processing
- Using batch records or absolute test methods Test
- failure filter integrity
following flowchart can help in understanding the classification of deviations
DEVIATION MANAGEMENT PROCESS FLOW
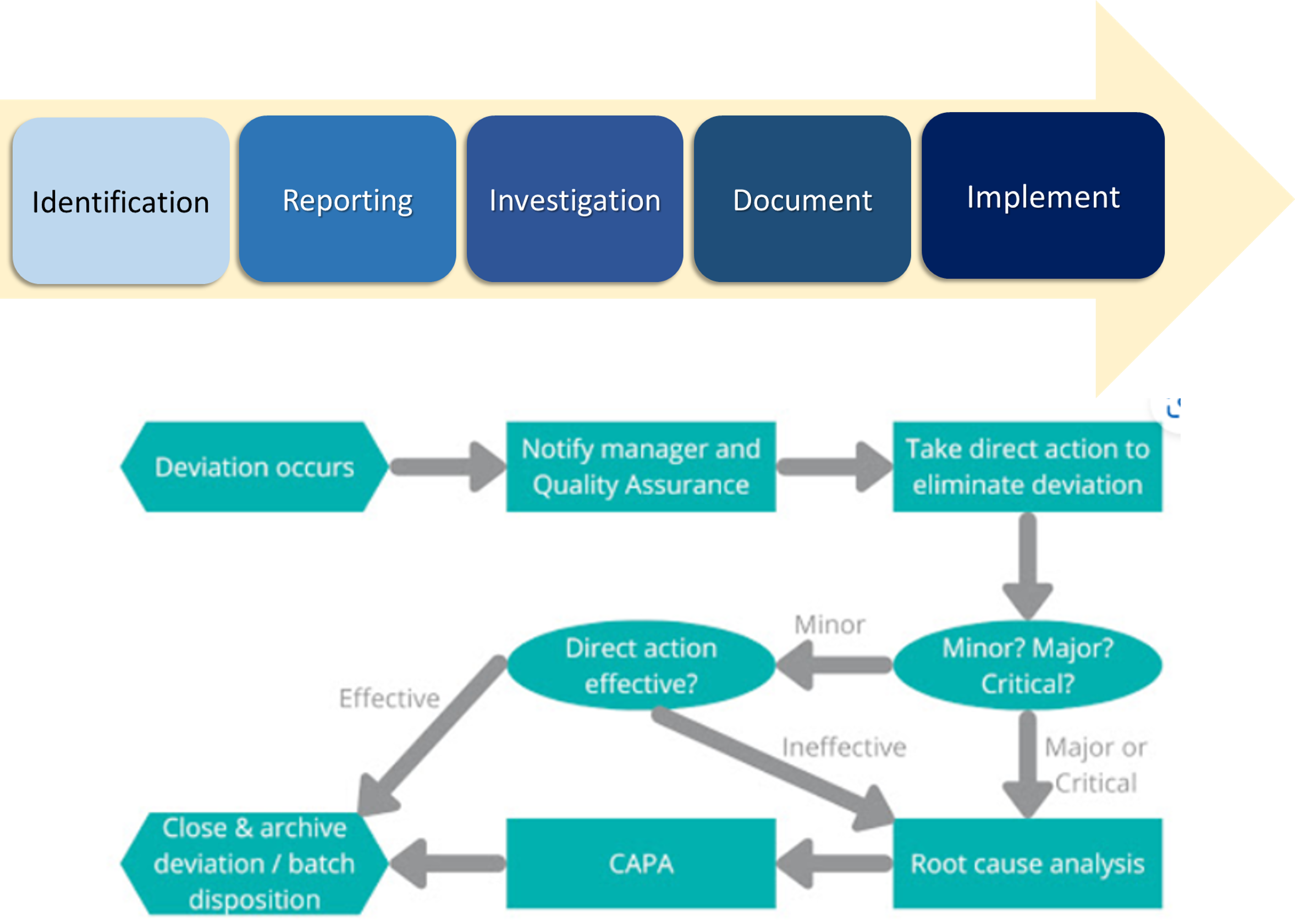
- Identification
- First of all, a clear description of the deviation that occurs
- All deviations / deviations must be registered and handled properly by the team responsible for quality risk management (Quality Risk Management)
- Collect all relevant information then categorize the type of deviation (Minor, Major, Critical).
- Registration uses a specific number:
- DN/YYYY/ZZZ
- DN: Deviation Number
- YYYY: Year (such as: 2022)
- ZZZ : Serial Number
- Reporting
- Organizations must be able to track all deviations that occur and have a policy to report them immediately if deviations occur.
- Deviations that occur are recorded and reported by the department where the deviation occurs.
- All personnel involved in handling deviations must provide all relevant information to assist investigations.
- Investigation
Failure Analysis
analysis is an investigation conducted to determine the cause of failure of a particular process/testing/product to prevent recurrence in the future.
Purpose
- Knowing how the process / test / product they will be. failed (Failure Mode).
- Where do product failures occur? (Failure Site).
- Identification of the physical phenomena involved in the failure (Failure Mechanism)..
- Determine the root of the problem (trigger points that caused the failure).
- Recommend Failure Prevention Methods (CAPA).
- Once deviations are identified and reported determine what is at the root of the problem.
- If the deviation is categorized as a critical deviation “Root Cause Analysis” needed.
- The relevant department cooperates with Quality Assurance who will carry out the "Root Cause Analysis".
- If the deviation is categorized as “Incident” or “Minor” “Root Cause Analysis” is not required.
- After “Root-Cause Analysis” is completed, the next step is Plan & create CAPA.
- After the “Quality Assurance” section approves the CAPA the department where the deviation occurs is responsible for implementing the CAPA to completion.
Investigation of a deviation should not be biased from the start.
- From a GMP perspective, the purpose of conducting a deviation investigation is not "to release" batches.
- The real purpose of a deviation investigation is to:
- Determine the "Root Cause" of the occurrence of the deviation.
- Implement appropriate and accurate corrective actions.
- Evaluation of the affected system, after repeated deviation patterns are recorded and analysed.

general problems in investigation

- Documentation
- After investigation of irregularities has been completed, document “Root Cause Analysis” and CAPA for tracking purposes..
- If deviation handling is handled electronically the audit trail needs to be printed for regulatory purposes.
- Audit Trail: a record that contains a chronology related to the actions taken, complete information about deviations, changes that occur, including “Root Cause Analysis” and CAPA.
- Implementation
After the CAPA is approved by the QA, implement the CAPA according to the agreed schedule/time.
System Linkage

DEVIATION HANDLING PROCEDURE
STEP #1: Describe the Deviation
Describe the deviation in detail.
Explain what, where, and when the deviation occurred. Stated simply and factually. It consists of two or three sentences that do not contain the proposed theory or conclusion.
Example, “On 21 Jun 2017 during the manufacture of Vanilla API Lot XYZ123, in manufacturing suite 401, it was observed that the material was clumping and not passing through the sieve on shaker machine #ABC1234.”
Application of the Investigation
Start the investigation by answering the following questions:
- What was found?
- Who was involved?
- When did the deviation event occur?
- Where did the deviation occur?
- How was the deviation discovered?
- How often does the deviation occur?
There is a BIG Difference between What Happens vs Why or How Something Happens
STEP # 2: Collection Data
collection is a critical step in finding the root cause of the problem
- Work objectively
- Collecting data without bias gives the best possibility to pinpoint the most likely root cause of the problem.
- The data taken include:
- Batch/lots number
- Environmental conditions where the deviation occurred
- Name of personnel involved
- Batch records, test results records
- Records or measurement results of raw materials
Important information in data collection
- Is the environment the cause of the problem?
- Are the working conditions appropriate?
- Are there problems at this stage of the process?
- Is there a problem with the facilities?
- Are there any equipment and raw material problems?
- Are the operating instructions clear?
- Are there communication problems with personnel or personnel training?
- Is there adequate supervision?
- Are there problems with methods, SOPS, work procedures or analytical methods?
- Has the process/procedure changed?
STEP # 3: Root Cause Analysis (“Root Cause”)
- Once the data is collected, a root cause analysis is carried out.
- There are several steps that can be taken to perform RCA.

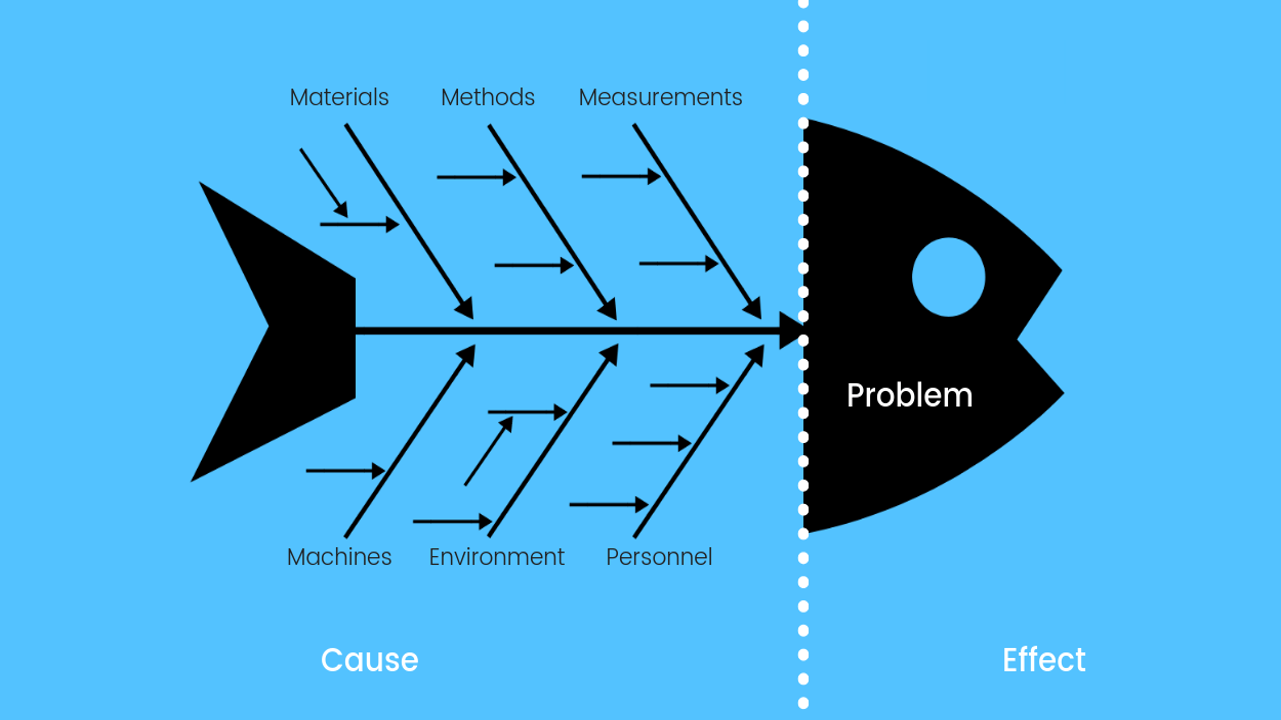
Cause Analysis
- Equipment
- Design, Capability
- Maintenance
- Operator Error
- Procedures
- No Procedure
- Wrong procedure used
- Procedure difficult to be used
- Training
- None
- When
- Effectiveness
- Management System
- Qualified Supervision
- Audits
- Feedback
- Corrective Action
- Planning
- Process Capability
- Adequate time to perform Task
- Standard
- Quality Control
- Inspection Required
- Inspection Performed
Personnel
- Identification each individual in the process
- Variation of abilities/competencies among personnel
- Evaluate the ability of employees to ensure that the steps of the procedure have been carried out correctly
Method
- Explain how the steps of the process are carried out.
- What are the specific requirements of the process?
- Are there critical-to-quality (CTQ) measures involved?
- Development methods related to the production process.
- There are critical changes that occur in the process that have an impact on product quality.
- Validation
- Trend data regarding deviations
Machinery & Equipment
- Identification of production tools and machines, production support facilities (HVAC, Water Purification System, Compressed Air, Steam) and test tools/instruments needed for the production process.
- Check calibration records
- Check machine maintenance records.
- Check the qualification report (KI/KO/KK)
- Check for the possibility of critical changes (Change Control)
- FAT/SAT
Raw Materials
- Check the Certificate of Analysis (CoA) of raw materials
- Check for possible “Mix-up”
- Check for possible “Cross-Contamination
- Check stability test report
- Check for possible damage to the primary packaging material.
Measuring Tools/Instruments
- What data is generated from the measuring tools/instruments used to evaluate product quality?
- Data Calibration
- Preventive Maintenance
- Qualification (KI/KO/KK)
- System Suitability
Environmental Monitoring
Identify conditions for operation:
- Temperature and humidity
- Pressure difference between rooms
- Measurement of the amount of contaminants (Viable & Non-viable)
- Air flow direction (laminar/ turbulent)
- Air change/hour
- Room Recovery
Tools for Rooting Problems
“5 Why’s
- Five Whys is a technique used to explore cause-and-effect relationships.
- Each "Why" question should form the basis for the next question.
- The final answer should indicate the cause of the process failure or defect.
- The purpose of the 5 whys is to determine the root cause of the defect or problem
- The investigator's tendency to stop at finding symptoms rather than getting to the root cause of the problem.
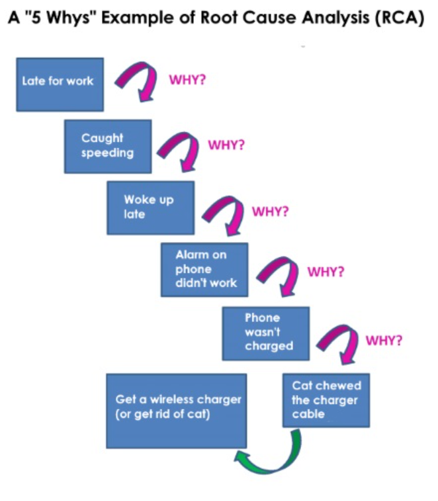
Other Tools for Rooting Problems
“5 Why’s
Example :

Other Tools for Rooting Problems
Pareto Analysis
Pareto analysis is based on the well-known Pareto Principle, which states that 20% of the work done will produce 80% of the results sought.
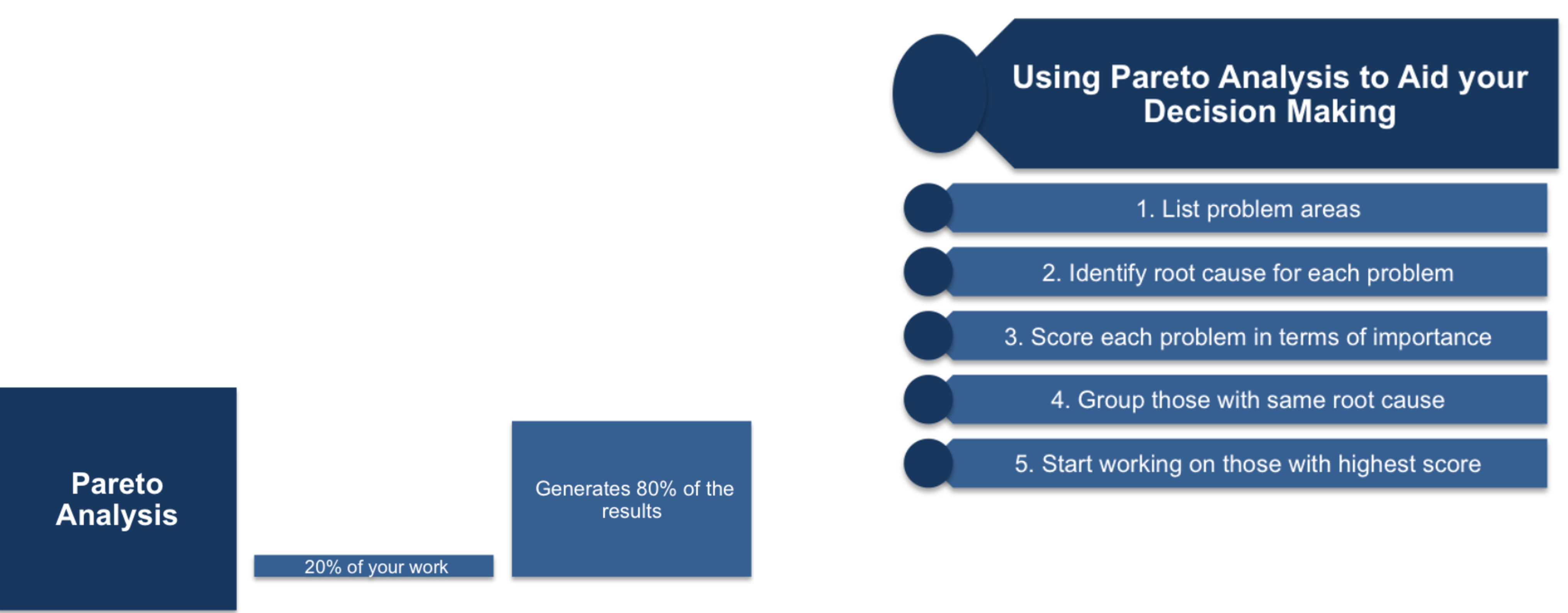


STEP # 4: Impact & Risk Assessment Analysis
- Once the root causes have been determined, carry out an impact and risk assessment.
- Consider the impact of the deviation on the product or quality system and how it relates to other products or quality systems.
- Many approaches and tools can be used to assess and manage risk assessments, such as:
- Failure mode and effects analysis (FMEA)
- Fault tree analysis (FTA)
- Hazard analysis and critical control points (HACCP)
- Hazard operability analysis (HAZOP)
- Preliminary hazard analysis (PHA)
Identify whether the risk assessment can answer the following questions:
-
- What could go wrong?
- What are the possible causes of error?
- What are the consequences (severity)?
- Next, rank the risks based on severity, likelihood, and detectability (RPN).
- Identify the appropriate risk severity level as Critical, Major, or Minor
- Determine the likelihood of the risk occurring as very likely, likely, or not likely.
- Determine the probability of detection by classifying the risk as easily found (easily found), somewhat hard to find (somewhat hard to find), or hard to find (hard to find).
- Based on the severity, risk frequency level, and probability of detection, determine the total risk level (RPN = Risk Priority Number).
- After determining the total risk level (RPN), the assessor team identifies which remediation strategy is best and designs specific actions (CAPA) to implement the agreed strategy.
STEP # 5: create CAPA
- Corrective Actions (CAs)
- An action taken to eliminate the root causes and symptoms of existing deviations or nonconformities and to prevent their recurrence.
- Reactive Actions to eliminate identified problems in products, services, or processes and address problem fixes directly
- Preventive Actions (PAs)
- are actions taken to eliminate potential causes of nonconformities, defects, or other undesirable situations and to prevent recurrence of errors .
- It is a PROACTIVE ACTION to avoid deviations and to prevent the same problem from recurring again.
- Immediate Action
-
Example :
- Product : Quarantine, Isolation, Destruction
- Machine/Equipment : Move from operating room and replace
- Process : process, testing is stopped.
- CAPA is taken to eliminate the root cause of deviation, and must be based on an objective and true investigation.
- Corrective actions (CAPA) must be approved by QA prior to implementation and their effectiveness verified and documented.
- If the CAPA is designed properly, the CAPA system will be able to prevent the occurrence of repeated deviations/errors
STEP # 6: Making Conclusions
The Conclusion section summarizes events, root causes (“Root Causes”), impacts, and risk assessments with reference to corrective or preventive actions..
STEP # 7: CAPA Effectiveness Check (PE)
- If a CAPA is required, an effectiveness check needs to be carried out.
- EC's goal is to develop internal monitoring that will generate metrics to evaluate how successfully event investigations are being managed and how capable CAPA can adequately address the root causes of irregularities.
This includes:
- Evaluating whether the same deviation occurs again in a given time period.
- How to continuously improve what can prevent the repetition of the same deviation.
- There are a number of deviations caused by lack of employee training
Evaluation of CAPA Effectiveness includes:
- CAPA completed and implemented on time
- CAPA is effective in reducing the occurrence of the same error/deviation
- Check whether CAPA has been implemented correctly.
- Determine the data sources needed for effective
- the evaluation period
- Determine the success parameters (Success Criteria)
- Evaluation of the effectiveness of the CAPA is carried out within 60 days of the implementation of the CAPA.
Study Case
Product of Vit C Effervescent Tablets, pack in strip, Batch No. : DI-120817, Manufactured in : August 2017, in November 2018 were found discolored in Bandung (20 Strips), Bogor (120 strips), Samarinda (80 pcs), West Papua (30 Strips) and Palembang (25 strips).
Prepare a Failure Investigation Report to identify the root-cause and to find the Corrective and Preventive Actions to prevent recurring this event.
Formulation
Effervescent consist of a soluble organic acid and an alkali metal carbonate salt, one of which is often the API. Carbon dioxide is formed if this mixture comes into contact with water.

Material Handling
- The primary material used in the manufacture of effervescent is relatively hygroscopic, it absorbs moisture from the air. This must be prevented because it will initiate the effervescent reaction.
- The principle strategies used to overcome this problem is a completely closed material handling system, which includes intermediate bulk containers (IBCs), docking stations and split valve technology
- The ventilating air must contain a sufficiently low moisture content.
- The material fed into the presses has to have properties that prevent segregation and ensure homogeneous filling of the dies, to produce tablets of equal weight. The most common approach to achieving materials with these characteristics is to granulate the raw materials.
- Dry methods, such as slugging, direct compression and roller compaction are regularly used to produce solid dosage forms. These are the preferred methods of producing effervescent because no liquid is involved, no additional drying step is required. However, the main argument against the use of dry methods is the need for expensive excipients.
- Wet Granulation
- For the wet granulation process, two separate granulation steps are run (one each for the alkaline and the acid components) with a subsequent dry blending step
- Proses pencampuran dapat menjadi langkah kritis dan dapat mempengaruhi homogenitas tablet karena tidak semua bahan-bahan tersebut diikat menjadi satu butiran seperti dalam proses granulasi basah konvensional.
- The effervescent reaction is only started if the materials come into contact with water - and not if they come into contact with organic solvents
- Organic solvents offers a number of advantages, which originate from the lower heat of evaporation when compared with water high throughput
- The only disadvantage of this method is the need for more complex equipment to handle the fluids, a complex system for exhaust gas treatment is required. This would not be the case in single pot dryer as only the organic vapor has to be handled.
- Fluid Bed Spray Granulation
- Fluid bed spray granulation is a unique process in which granulation and drying take place simultaneously. This ensures a constant low moisture level, limiting the pre-effervescent reaction to a minimum.
- It is easy to achieve a very low final moisture level for storage.
- Lubricant
- Its function is to improve the flow of the material, which is extremely important because the dies of a tablet press are filled by volume. A second function is to prevent the tablet sticking to the punch faces or to the walls of the dies.
- Magnesium stearate should not be used because they are insoluble in water and, consequently, a film will form on top of the water after the tablet has dissolved.
- Tablet Compression
- The compression of effervescent tablets is different from the compression of normal tablets.
- For long-term storage, a very low moisture content is required - typically less than 0.3% water compared with approximately 2% for traditional tablets.
- Effervescent tablets are usually quite large, which often leads to insufficient tablet hardness and consequently broken or damaged tablets
- Packaging
- Packaging After the material has been pressed into tablets, the surface area of the material has been significantly reduced, which means that the rate at which moisture is absorbed from the air has also been reduced.
- Aluminum, which has low water permeability, is used instead of standard polymer blister materials.
- If ten or more individual tablets are packed into one tube, very dry air can be added but as the patient opens the tube to take out the first tablet, ambient moist air will enter and destroy the remaining effervescent tablets.
References
- Guidance for Industry Quality Systems Approach to Pharmaceutical CGMP Regulations , FDA, Sep 2006
- PIC/S guide to good practices, for the preparation of medicinal products in healthcare establishments, PE-0103, Oct 2008
- FailureAnalysisandPrevention,Vol11,ASMHandbook,ASMInternational, 1986, p 15–46
- Deviation Handling and Quality Risk Management, World Health Organization (WHO), Geneva, Switzerland , July, 2013
